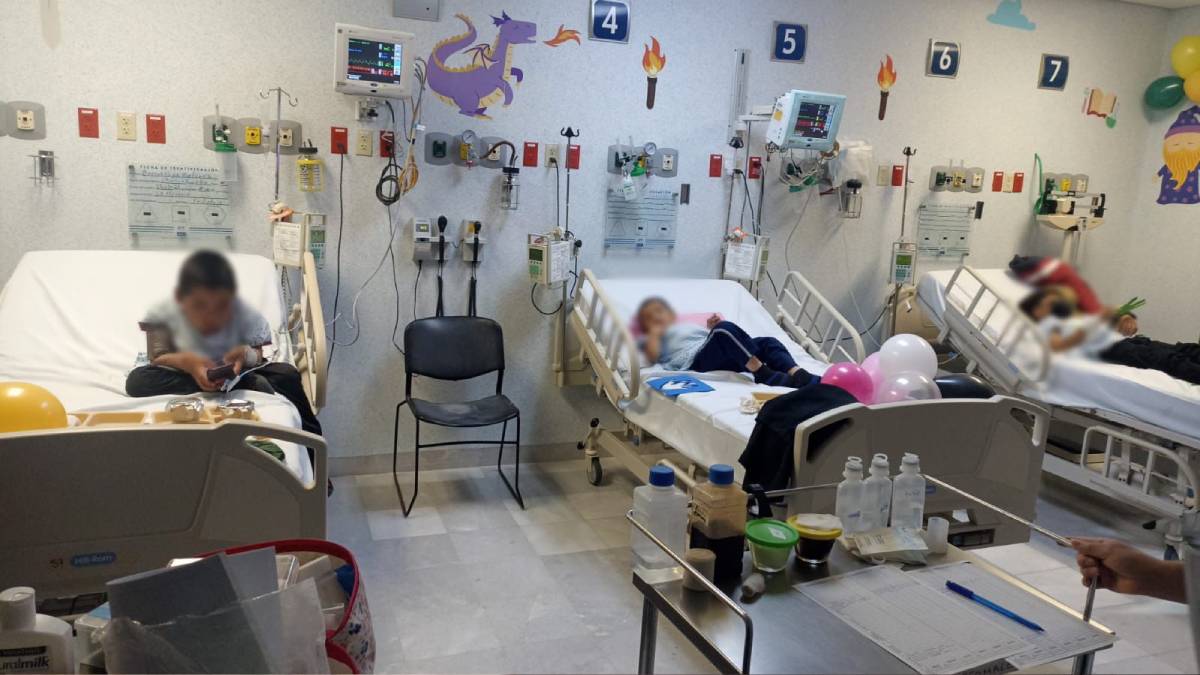
Guanajuato.- José Baldomero y Antonio Landa representan la lucha diaria de quienes viven con enfermedades raras en Guanajuato. A pesar de enfrentar padecimientos distintos, ambos comparten historias marcadas por el dolor, la resiliencia y la urgencia de recibir un tratamiento especializado para recuperar su calidad de vida.
A sus 22 años, Baldomero convive con la “piel de mariposa”, una condición genética que vuelve su cuerpo tan frágil que cualquier contacto le provoca heridas.
Tras una década de diagnósticos erróneos y falta de atención, hoy encuentra refugio en el Hospital Pediátrico de León, manteniendo firme el sueño de viajar con su familia para conocer el mar.
Por otro lado, la vida de Antonio, un músico de 33 años, cambió drásticamente en solo un mes debido al síndrome de Guillain-Barré.
Lo que inició como un supuesto cuadro de estrés escaló hasta arrebatarle el habla, la vista y la movilidad, obligando a su familia a buscar ayuda económica urgente para costear las terapias que podrían regresarle su autonomía.
José Baldomero: El joven con piel de mariposa y voluntad de acero
José Baldomero Jiménez Tovar tiene 22 años, es el segundo de cuatro hijos, le gustan mucho los videojuegos y sueña con viajar con su familia a la playa para conocer el mar; a pesar de que vive con epidermólisis bullosa distrófica, también conocida como “Piel de mariposa”, expresó que sí se puede seguir adelante.
“Me gusta jugar videojuegos y tratar de apoyar a mi familia y a mis papás en lo que pueda porque obviamente no puedo hacer mucho, pero intento ayudar un poco en las cosas del hogar”.
También tiene un mensaje:
Decirles a los niños y jóvenes que viven como yo que sigan adelante, sí se puede seguir. Es un poco difícil y doloroso, pero nos tocó así a nosotros y tenemos que seguir adelante de alguna u otra manera. Me gustaría ir de viaje con mi familia a una playa para conocer el mar”, compartió a AM.
El pasado miércoles José Baldomero, acompañado de su mamá, Alma Delia Tovar, acudió al Hospital de Especialidades Pediátrico de León para recibir material de curación y suplementos alimenticios.
Ambos, entrevistados por AM, dijeron estar agradecidos con la dermatóloga pediatra, Dulce María Ortiz Solís, quien junto con otros médicos especialistas atienden a los pacientes con esta enfermedad rara o poco frecuente en Guanajuato.
Me trata muy bien la doctora Dulce, es muy buena doctora y siento bonito venir aquí al Hospital Pediátrico por la atención que recibo por parte de los doctores. Estoy muy agradecido con la doctora Dulce porque es quien nos está apoyando en esta situación”, expresó el joven originario de San Miguel de Allende.
“Es una lección de vida muy fuerte”
Alma Delia Tovar, mamá de José Baldomero, compartió que durante 10 años su hijo no recibió atención médica, pues a pesar de tener el diagnóstico, los pediatras y los médicos a los que recurrieron no supieron cómo tratar su enfermedad.
Fue hasta un año después de que abrió el Hospital de Especialidades Pediátrico de León, en 2016, cuando desde un centro de salud de San Miguel de Allende, Baldomero fue referido a esta unidad.
“Hemos encontrado como un refugio en el Hospital Pediátrico y con la doctora Dulce Ortiz porque ella nos ha sabido explicar de la enfermedad, de cómo sobrellevarla y cualquier situación ella está siempre para atendernos”.
Él nació en Celaya y regresamos con el pediatra que lo atendió primero y nos decía que no se podía hacer nada, que no había medicamentos para esto y que lo único que había que hacer era esperar a que falleciera porque iba a llegar un momento en que se iba a infectar”, contó.
Alma recordó que durante los primeros días de vida de Baldomero pensaron que iba a fallecer; tras noches en vela, ella y su esposo aprendieron sin ayuda médica cómo cuidarlo y curar sus heridas.
“Pasamos casi 10 años sin que lo revisara un médico porque íbamos con algunos doctores, incluso fuimos al Hospital Infantil de México Federico Gómez y no tenían el conocimiento de la enfermedad. Al grado de que llegaron a lastimarlo”.
“Le poníamos en sus heridas algodón porque veíamos que le absorbía, pero la doctora Dulce nos explicó que había que ponerle gasas y ungüentos para que no se infecte y se fueron haciendo más pequeñas sus heridas. Nos ha enseñado mucho la doctora”, contó.
Dejó de estudiar
Los papás de Baldomero sabían cuál era su enfermedad, sin embargo no que hay tres tipos; su hijo tiene el tipo más grave, que es la epidermólisis bullosa distrófica.
Debido a esta condición de salud y por el bullying que vivió en sexto de primaria, el joven ya no estudió.
“Empezaron a hacerle notar por sus manos y a ponerle apodos, por eso decidimos no inscribirlo a la secundaria y fue un error porque debimos seguir con su educación. Ha estado con nosotros todo el tiempo”.
“Al inicio no sabes qué hacer, uno piensa lo peor y no debe ser así, hay que darles la oportunidad de desarrollarse más. La doctora Dulce es como un ángel para nosotros y para todos los otros niños con esta enfermedad. Esta es una lección de vida muy fuerte”, expresó Alma Delia.
Atiende Pediátrico a 18 con “Piel de mariposa”

Actualmente, el Hospital de Especialidades Pediátrico de León brinda atención a 18 pacientes con epidermólisis bullosa, también conocida como “Piel de cristal” o “Piel de mariposa”, algunos ya mayores de edad.
Así lo informó la dermatóloga pediatra Dulce María de las Mercedes Ortiz Solís, quien labora en dicha unidad de tercer nivel donde se diagnostican y tratan enfermedades raras o poco frecuentes.
Me encargo de ver una enfermedad que se llama epidermólisis bullosa y que se conoce como ‘piel mariposa’ porque se compara con la fragilidad de las alas de las mariposas. Son pacientes que nacen ampollados y que viven prácticamente toda su vida así”.
Explicó que esta enfermedad se clasifica en tres tipos: la simple, la distrófica y la de unión. Lo que pasa es que a la persona le faltan ciertas proteínas en la unión dermoepidérmica (las dos capas más externas de la piel).
Esto hace que la piel se desprenda, lo que ocasiona la formación de las ampollas, por lo que el tipo de proteína faltante definirá el tipo de epidermólisis que tendrá la persona.
Ortiz Solís agregó que esta enfermedad es hereditaria, pero aclaró que hay casos que ocurren sin que haya antecedentes de este padecimiento en la familia.
Hay dos tipos de forma de herencia, la herencia dominante y la herencia recesiva. Cuando es dominante quiere decir que ya existe el antecedente en la familia o que alguien tiene el gen alterado”.
“Y cuando es herencia recesiva es más severa, pero también por ahí anda el gen en la familia, pero no es de madre a hijo, sino que puede ser de los abuelos o de una generación anterior”, explicó.
También hay una tercera forma de herencia, conocida como la herencia de novo, que ocurre cuando no hay ningún antecedente en la familia y se dio una mutación durante el embarazo.
¿Cómo tratarla?
La epidermólisis bullosa distrófica es uno de los tipos más graves; la esperanza de vida de las personas que la tienen es de entre 25 y 30 años debido a las complicaciones que se desarrollan.
“Son pacientes que nacen con ampollas y es muy difícil tratarlos porque ponerles un pañal les irrita, si se cargan de forma inadecuada también se ampollan. A estos pacientes les falta el colágeno tipo 7”.
No tienen problemas neurológicos, aprenden bien y pueden ir a la escuela, pero muchas veces dejan de ir porque necesitan muchos cuidados”, lamentó la doctora Ortiz.
Enfatizó que la piel de las heridas cicatriza pero no se adhiere y, al cicatrizar, en el caso de las manos se van pegando los dedos hasta que quedan solo los muñones, condición que requiere adaptaciones en el hogar.
“Prácticamente quedan sin dedos y eso es muy difícil. Conforme van creciendo hay que enseñarles a comer porque no pueden comer alimentos sólidos que les puedan irritar el esófago; llega un momento en el que pueden comer solamente líquidos”.
“Desafortunadamente por esta condición mueren por inanición entre los 25 y 30 años. Son personas que no crecen, que tienen anemia y muchas complicaciones. Los pacientes distróficos viven con cicatrices”, expresó.
Llegaron al Pediátrico tras ser desahuciados
Aunque no precisó cuántos, la doctora Dulce Ortiz aseguró que la mayoría de los pacientes atendidos en el Hospital Pediátrico llegaron cuando tenían entre 8 y 12 años debido a que fueron desahuciados al nacer.
Les dijeron que se iban a morir y los papás como pudieron los cuidaron. Cuando les hicimos el diagnóstico las mamás estaban agradecidas de saber qué tienen sus hijos”.
“Son pacientes que requieren atención psicológica y que son atendidos por especialistas en ortopedia, odontología, genética, gastroenterología, nutrición y la Clínica de Heridas”, resaltó.
Detalló que desde su apertura, en enero del 2015, a la fecha, el hospital tiene registro de 23 pacientes con epidermólisis bullosa, pero activos hay 18 debido a que cinco ya fallecieron.
Y si los pacientes tienen IMSS o ISSSTE los seguimos atendiendo porque muchas veces los compañeros médicos de estas instituciones nos consultan, no se les niega la atención”.
“Muchas gracias a las autoridades de Salud del Estado porque hacen posible que esto pase y podamos seguir atendiendo a pacientes que incluso ya son adultos”, destacó.
Brinda APEA apoyos
En Guanajuato, la Asociación Pro Epidermólisis Ampollosa (APEA), conformada por padres de familia, realiza la venta de mazapanes y otras actividades para recaudar fondos y recibir donativos de material de curación.
Somos un brazo de la Fundación Debra, que es una asociación internacional que se dedica a la investigación y al apoyo de estos pacientes. Debra México se fundó en Monterrey hace 28 años”, apuntó la doctora Dulce Ortiz.
Agregó que también se apoya a los pacientes de otros municipios con los gastos de transporte para sus consultas y, cuando no pueden acudir a León, se les envía el material de curación hasta su hogar.
En cifras
La Secretaría de Salud de Guanajuato (SSG) tiene cinco hospitales capacitados para el tratamiento de enfermedades raras en San Luis de la Paz, Celaya, San Miguel Allende, Acámbaro y León.
Hasta el cierre de diciembre de 2025, se atendían 22 pacientes con diversos tipos de enfermedades raras, siendo la más frecuente la mucopolisacaridosis; 11 son de Guanajuato, uno de San Luis Potosí y 10 de Michoacán.
Antonio Landa: El joven músico que lucha contra el síndrome de Guillain-Barré

Lo que comenzó como síntomas leves, hoy es un caso clínico grave para Antonio Landa Dávila, quien en cuestión de un mes ha dejado de caminar, de hablar y perdió también la vista debido al síndrome de Guillain-Barré.
Antonio Landa es un joven de 33 años, diseñador y músico apasionado por el género punk; hoy enfrenta la lucha contra esta enfermedad autoinmune que afecta principalmente el sistema nervioso periférico.
De acuerdo con la familia de Antonio, todo comenzó en diciembre, cuando empezó a sentir adormecimiento en parte de la cara y brazos, además de las piernas, lo que en ocasiones le dificultaba caminar.
En un inicio, se creyó que eran síntomas de estrés, ya que, aunque acudió tanto al Seguro Social como a médicos particulares, los análisis no detectaron nada anormal, por lo que supusieron que tendría que ver con cuadros de ansiedad.
Desde hace un mes, Antonio tuvo que mudarse con su familia nuevamente, ya que perdió la movilidad; en cuestión de semanas dejó de ver e incluso de hablar, pese a que ya estaba recibiendo tratamiento psiquiátrico bajo un diagnóstico inicial erróneo.
Diagnóstico oficial
Fue a mediados de la semana pasada cuando Antonio tuvo que ser internado en el IMSS de San Francisco del Rincón ante las sospechas de un tumor, por lo que se le envió a León para realizarle una resonancia magnética y una tomografía.
Si bien los resultados descartaron tumores, la familia de Antonio aseguró que los médicos le detectaron formalmente el síndrome de Guillain-Barré, el cual terminó siendo el responsable de sus síntomas más agudos.
Por ello, la familia de Antonio pide ayuda para el tratamiento, pues el joven vivía solo y, debido a su padecimiento, tuvo que dejar de trabajar, mientras que la situación económica dificulta la adquisición de medicamentos, terapias y estudios.
A través de la página de GoFundMe, los familiares iniciaron una recaudación de fondos; también solicitaron apoyo moral o espiritual, asegurando que “cada ayuda es bienvenida”.
¿Qué es el síndrome de Guillain-Barré?
- Es una afección rara en la que el sistema inmunitario de una persona ataca los nervios periféricos.
- Puede afectar a personas de todas las edades, pero es más frecuente en adultos y en hombres.
- La mayoría de las personas se recuperan totalmente, incluso en los casos más graves de la enfermedad.
- Los casos agudos son raros, pero pueden provocar una parálisis casi total y problemas para respirar.
- El síndrome puede ser mortal. Los pacientes necesitan tratamiento e inmunoterapia de forma inmediata.
AAK
Acompáñanos en un recorrido por la historia de León. Recibe en tu correo relatos sobre personajes, barrios, tradiciones y momentos clave, que celebran la identidad leonesa, en el marco de los 450 años de nuestra ciudad.